Welch Lab Research Focus
Our research aims to address fundamental problems in both biomedical research and computer science by developing new tools tailored to rapidly emerging high-throughput sequencing technologies. Broadly, we seek to understand what genes define the complement of cell types and cell states within healthy tissue, how cells differentiate to their final fates, and how dysregulation of genes within specific cell types contributes to human disease. As computational method developers, we seek to both employ and advance the methods of machine learning, particularly for unsupervised analysis of high-dimensional data.
Most recently, we have focused on developing open-source software for the processing, analysis, and modeling of single-cell sequencing data. Key contributions in this area include LIGER, a general approach for integrating single-cell transcriptomic, epigenomic and spatial transcriptomic data; online iNMF, a scalable and iterative algorithm for single-cell data integration; and MultiVelo, a tool for modeling cell fate transitions from single-cell multi-omic data. We have applied these methods in collaboration with biological scientists to study stem cell differentiation, somatic cell reprogramming, and the mammalian brain.
Research highlights
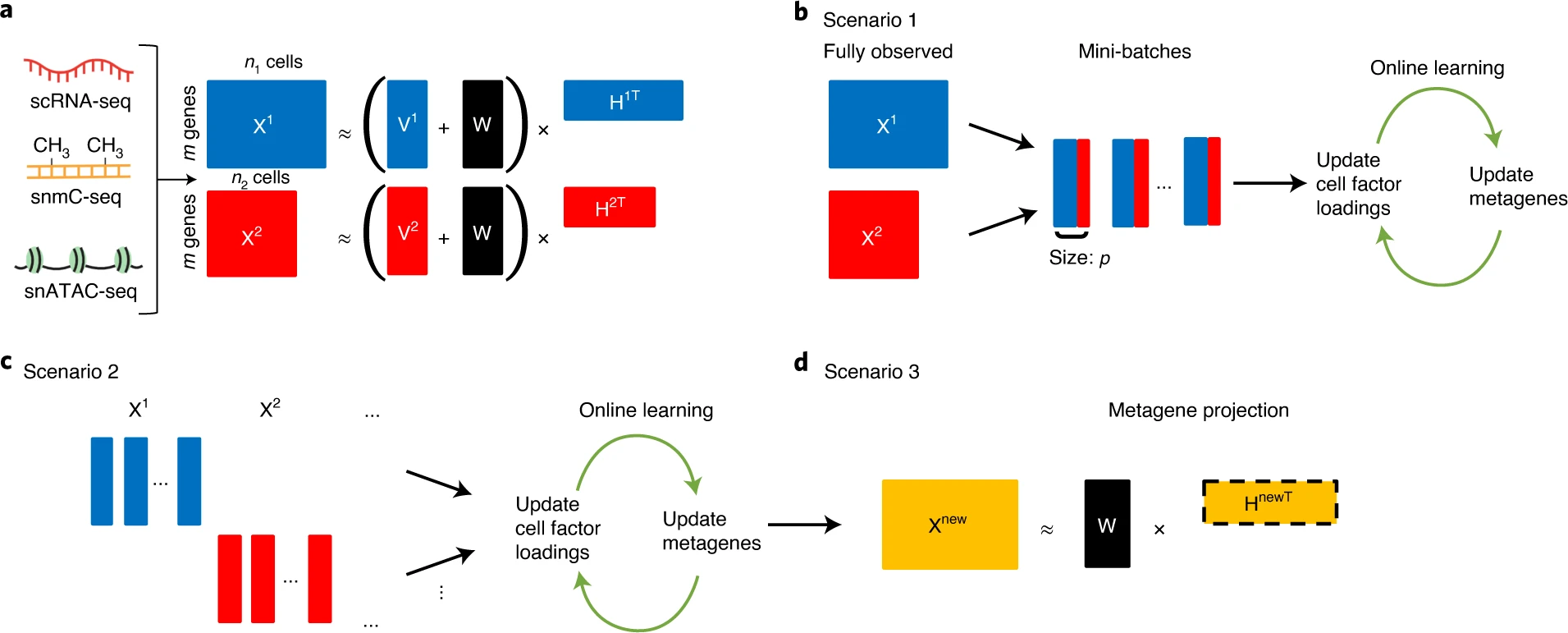
In papers published in Cell and Nature Biotechnology, we developed LIGER and online iNMF. These approaches allow integration of single-cell transcriptomic, epigenomic and spatial transcriptomic datasets from different single cells.
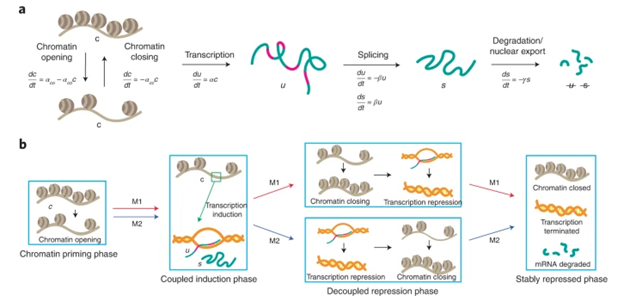
We developed MultiVelo, VeloVAE, and TopoVelo--approaches for modeling sequential transcriptomic and epigenomic changes in differentiating cells using differential equations.
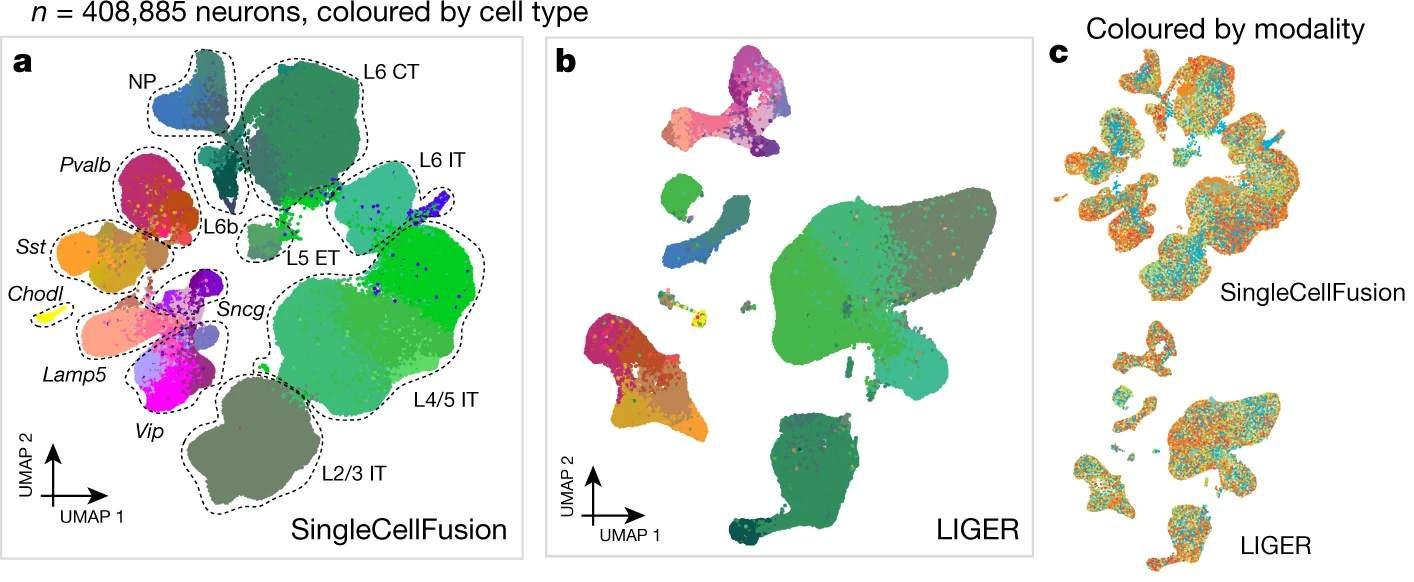
As part of the BRAIN Initiative Cell Atlas Network, we are integrating single-cell transcriptomic, epigenomic, and spatial datasets to characterize the cell types in the mammalian brain.
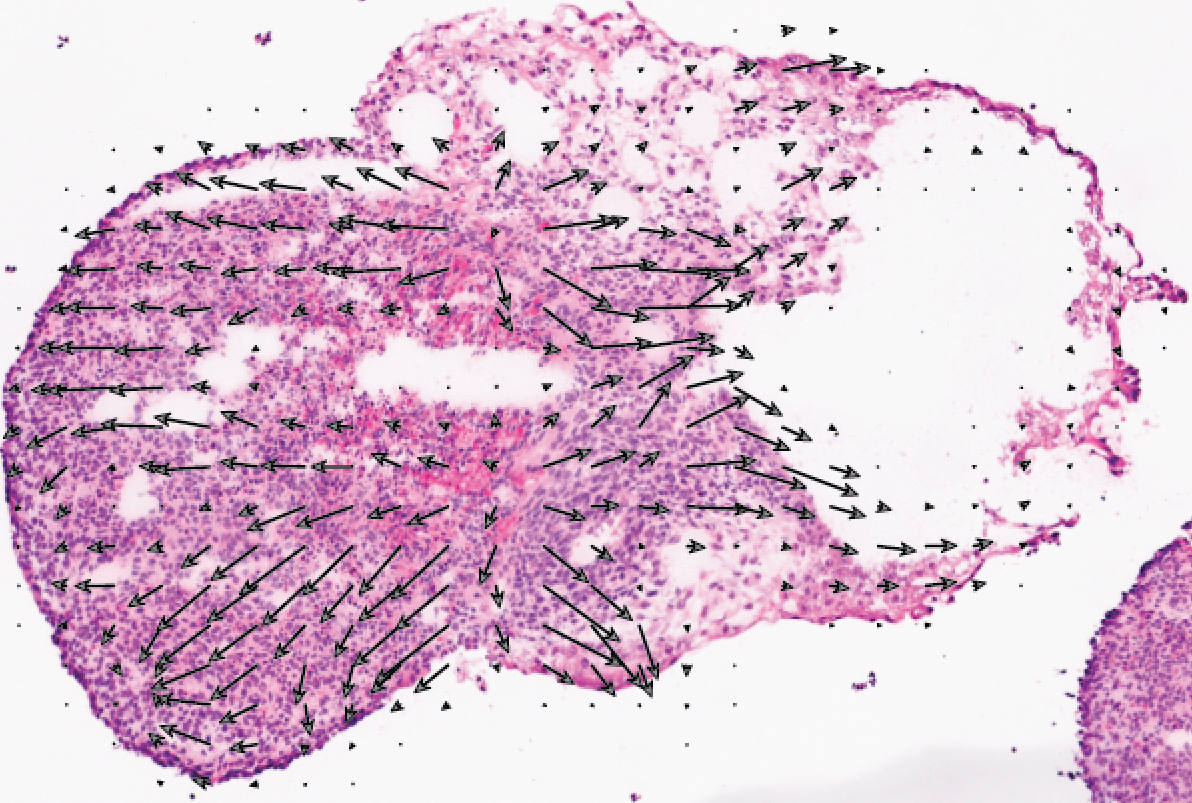
Using the algorithms we have developed, we are investigating in vitro models of development and disease, including directed differentiation and organoid models.