Simplex Analysis on Human Hematopoietic Stem And Progenitor Cells (HSPCs)
Yichen Wang, Chen Li
04/23/2025
Source:vignettes/articles/hspc_example.Rmd
hspc_example.RmdIntroduction
CytoSimplex visualizes the similarity between single cells and the centroids of selected cell clusters, which serve as the vertices standing for terminal states of differentiation. The similarity between each cell to all vertices is normalized to a unit sum to be plot as the barycentric coordinates. CytoSimplex makes use of the RNA velocity analysis result, presented as a cell-cell transition graph, to further visualize the differentiation potential from cells to each terminal. For detailed tutorial on the usage of the package, please refer to CytoSimpelx vignette.
HSPC dataset
In this real-life example tutorial, we present the simplex analysis on the single-nucleus RNA and ATAC multiome dataset from our published study of HSPC (Chen Li et al., Nat. Biotechnol, 2022). We adopt the RNA data for making the simplex plots, and adding the RNA velocity computed with MultiVelo for showing the differentiation potential. For detail of the RNA velocity analysis, please refer to the original publication.
The necessary input information for creating CytoSimplex plots includes the RNA raw count matrix, the annotation and the cell-cell transition graph from RNA velocity analysis.
The RNA raw count data is provided in a LOOM file, which is the
output of velocyto for the original
study. Function readVelocytoLoom() is provided specifically
for loading the raw count data from velocyto output LOOM
file.
- LOOM file: GSM6403410_3423-MV-2_gex_possorted_bam_ICXFB.loom.gz
- 36,601 features by 14,895 cells
- Decompress the file before loading
Users can also have their own raw count matrix imported through various approaches depending on the real format:
- For 10X cellranger output,
Seurat::Read10X()is the most handy and commonly installed tool. - For other types of loom files, please find loomR Tutorial for a clearly guided general procedure.
- For H5 files, most likely also of 10X cellranger output format,
Seurat::Read10X_h5()is the function to use.
The velocity analyses most often are performed in Python and organized using AnnData object, which is stored in H5AD files on disk. This tutorial provides the H5AD file of Chen’s published analysis, where annotation and MultiVelo results are available. CytoSimplex also provides convenience functions for extracting relevant H5AD data without having to link to a Python binary using reticulate. For the detailed guide for performing MultiVelo analysis, please refer to the MultiVelo documentation.
- H5AD file: hspc-7th-day-uninfected_multivelo_result.h5ad
-
adata.shape: 11,605 cells by 936 features -
adata.obs['leiden']: cell type annotation, containing 11 clusters. -
adata.uns['velo_s_norm_graph']: The RNA velocity cell-cell transition graph, 11,605 cells by 11,605 cells.
-
Please note that filtering might have been applied in the original analysis, so it is important to make sure the cell/nucleus identifiers (i.e. barcodes) are in a identically matching manner between the imported raw count matrix and the information extracted from the H5AD file.
library(CytoSimplex)
# For combining multiple plots / can use `cowplot` as an alternative
library(patchwork)
# For plot output customization
library(ggplot2)
# Load the raw count data from Velocyto output LOOM file
rna <- readVelocytoLoom("GSM6403410_3423-MV-2_gex_possorted_bam_ICXFB.loom")
# Load annotation and velocity data from H5AD file
adataPath <- "hspc-7th-day-uninfected_multivelo_result.h5ad"
barcodes <- readH5ADObsNames(adataPath)
annotation <- readH5ADObsVar(adataPath, "leiden")
velo <- readH5ADUnsSpMat(adataPath, "velo_s_norm_graph")
dimnames(velo) <- list(barcodes, barcodes)
# Size overview of the data
sprintf("rna: %s class, %d x %d", class(rna), nrow(rna), ncol(rna))
sprintf("annotation: %s class, %d values, %d levels", class(annotation), length(annotation), nlevels(annotation))
sprintf("velo: %s class, %d x %d", class(velo), nrow(velo), ncol(velo))## [1] "rna: dgCMatrix class, 36601 x 14895"
## [1] "annotation: factor class, 11605 values, 11 levels"
## [1] "velo: dgCMatrix class, 11605 x 11605"Here we do some extra preprocessing to make sure that the cell IDs match between the raw count and velocity analysis that come from separate sources.
# Check the format of cell IDs
sprintf("First 2 barcodes from matrix: %s", paste(colnames(rna)[1:2], collapse = ", "))
sprintf("First 2 barcodes from H5AD: %s", paste(barcodes[1:2], collapse = ", "))
# Simply force the barcodes in the raw count matrix to match the H5AD barcodes
colnames(rna) <- gsub("^gex_possorted_bam_ICXFB:", "", colnames(rna))
colnames(rna) <- gsub("x$", "-1", colnames(rna))
sprintf("First 2 modified barcodes from matrix: %s", paste(colnames(rna)[1:2], collapse = ", "))
# Check the matching
sprintf("Whether all cells in H5AD are found in matrix: %s", all(barcodes %in% colnames(rna)))
# Expected to be TRUE
# Then subset the matrix to the same set of cells in the same order
rna <- rna[, barcodes]
# Deduplicate the rownames (optional to most of other cases)
rownames(rna) <- make.unique(rownames(rna))
sprintf("Dimension of the subset matrix: %d x %d", nrow(rna), ncol(rna))## [1] "First 2 barcodes from matrix: gex_possorted_bam_ICXFB:AAAGCGGGTGAGACTCx, gex_possorted_bam_ICXFB:AAACCGAAGGATCCGCx"
## [1] "First 2 barcodes from H5AD: AAACAGCCAAACCTTG-1, AAACAGCCACCCTCAC-1"
## [1] "First 2 modified barcodes from matrix: AAAGCGGGTGAGACTC-1, AAACCGAAGGATCCGC-1"
## [1] "Whether all cells in H5AD are found in matrix: TRUE"
## [1] "Dimension of the subset matrix: 36601 x 11605"Set up the vertices
Based on prior knowledge from the original study, we select the clusters for progenitor megakaryocytes (PMKs), erythrocytes (ERs), and progenitor dendritic cells (PDCs) as the main three terminal cell fates for the ternary simplex analysis.
vt.tern <- list(
PMK = "Prog MK",
ER = "Erythrocyte",
PDC = "Prog DC"
)Select top features (optional but recommended)
For biological single-cell transcriptomics data of high dimensions, we recommend selecting the top 30 marker features of the terminal cell types for reducing the dimensionality and noise.
We implemented a fast Wilcoxon rank-sum test method specifically for
CytoSimplex workflow, which can be invoked with function
selectTopFeatures().
gene.tern <- selectTopFeatures(x = rna, clusterVar = annotation, vertices = vt.tern, nTop = 30)
print(gene.tern)## [1] "KLF1" "SLC40A1" "APOC1" "TFR2" "ANK1" "TFRC"
## [7] "S100A6" "KEL" "BLVRB" "RYR3" "EMP3" "CSF2RB"
## [13] "HDAC7" "S100A4" "MYH10" "ITGA2B" "HBD" "EPOR"
## [19] "TRIB2" "SPTA1" "PDCD4" "APOE" "LDB1" "HMGB1"
## [25] "ICAM4" "TSC22D1" "WNT5B" "KCNH2" "HSPG2" "CTNNBL1"
## [31] "IRF8" "LY86" "CD180" "TGFBI" "MPEG1" "CYBB"
## [37] "SAMHD1" "FGL2" "CD86" "CSF1R" "VCAN" "JAML"
## [43] "IGSF6" "ARL4C" "BLNK" "CCR2" "CLEC10A" "TIFAB"
## [49] "DTX4" "LILRB4" "SULF2" "AFF3" "CTSH" "GPR183"
## [55] "CXorf21" "CD74" "CES1" "SLAMF7" "SLC8A1" "RBM47"
## [61] "LTBP1" "MMRN1" "CMTM5" "THBS1" "MPIG6B" "ADCY6"
## [67] "LAT" "GP1BB" "ITGA6" "VWF" "ABCC4" "GP9"
## [73] "ITGA2B" "PROS1" "STOM" "F2R" "CKB" "MEST"
## [79] "DAAM1" "ITGB3" "GP1BA" "CTTNBP2" "CAMK1" "CLCN4"
## [85] "MYLK" "SLC37A1" "PLEK" "F2RL3" "C6orf223" "NFIB"Optionally, it is also tested that PCA as an alternative method for dimensionality reduction returns comparably good results. This will be demonstrated in the following sections.
Ternary plots reveal tri-lineage differentiation
plotTernary() shows sample similarity in a ternary
simplex – equilateral triangle. The closer a dot, a cell, is to one
vertex, the more similar the cell is to the cell cluster(s) the vertex
represents.
# Optional color settings to match the manuscript
hspc.colors <- c(
LMPP = "#714294",
HSC = "#CC131F",
MEP = "#78433A",
MPP = "#CD62A3",
Erythrocyte = "#1763A7",
GMP = "#F0691A",
`Prog MK` = "#FAAD65",
Granulocyte = "#249156",
`Prog DC` = "#9FB9E1",
`Prog B` = "#22B2C6",
Platelet = "#A8B250"
)
vt.tern.colors <- c(hspc.colors['Prog MK'],
hspc.colors['Erythrocyte'],
hspc.colors['Prog DC'])
# Create ternary plot
plotTernary(
x = rna, clusterVar = annotation, vertices = vt.tern,
features = gene.tern, veloGraph = velo,
dotColor = hspc.colors, labelColors = vt.tern.colors
)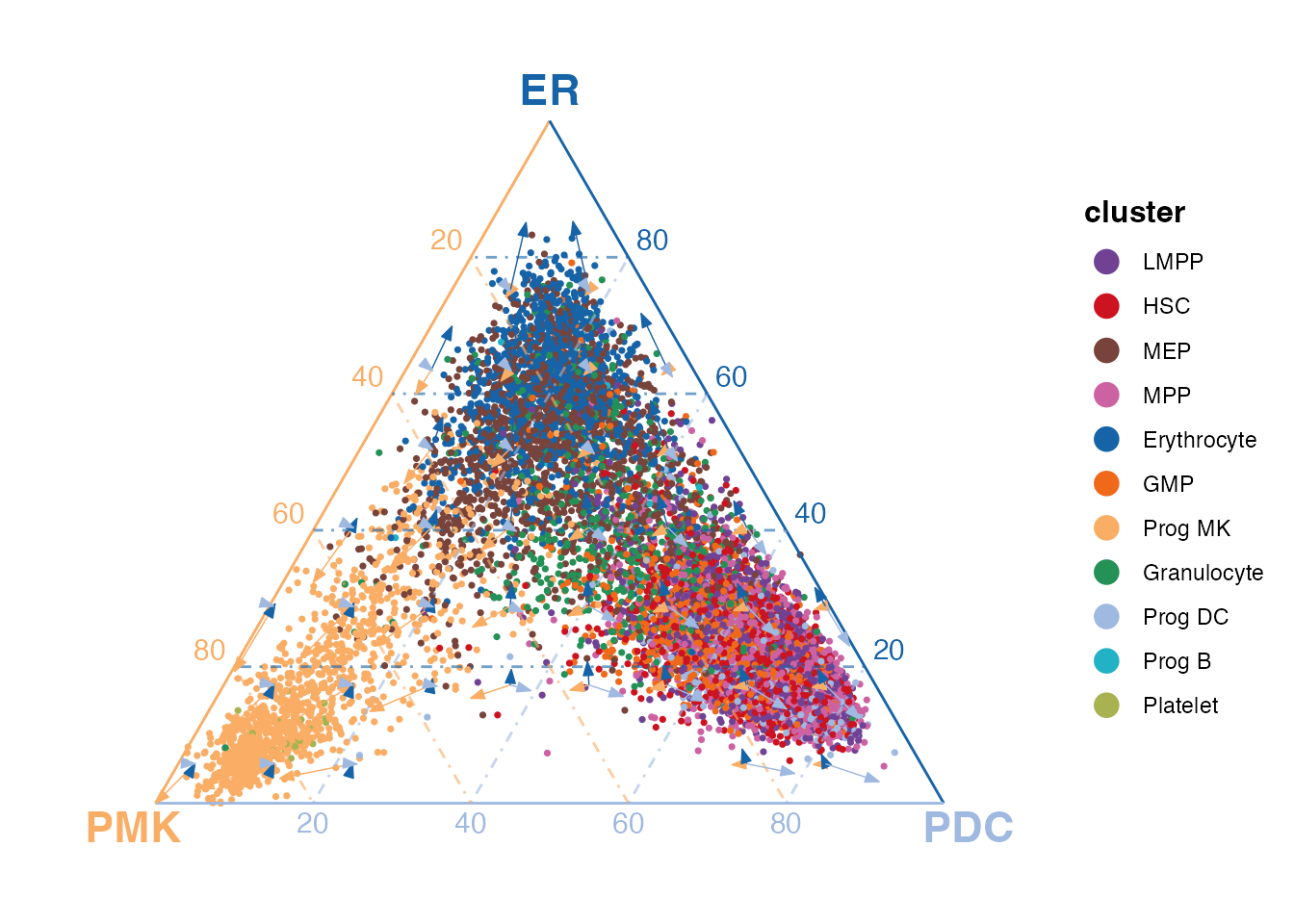
From the figure that includes all cells of the HSPC dataset, we can clearly tell that the cells belonging to the terminal clusters stays close to the corresponding vertices. Furthermore, the hematopoietic stem cells (HSCs) have high transcriptomic similarity to the PDCs, a group of lymphoid cells, while megakaryocyte-erythroid progenitors (MEPs) exhibit similarities to ER cells, which belong to the myeloid lineage.
Based on the prior knowledge of HSPCs, the cells are capable of differentiating into mainly the myeloid lineage and lymphoid lineage. Simply from the figure above, the observations are consistent with the known differentiation potential of HSPCs.
Below we show more detailed exploration within each cluster.
Interactive view for more details
Simply setting interactive = TRUE in the function call
will generate an interactive plot powered by plotly. Users can drag within the plot
region to zoom in and out for detailed view of local structure. Hovering
the mouse pointer on a dot or arrow shows detailed information. Clicking
on the a legend tag toggles the visibility of the corresponding cluster
or arrow.
plotTernary(
x = rna, clusterVar = annotation, vertices = vt.tern, features = gene.tern, veloGraph = velo,
dotColor = hspc.colors, labelColors = vt.tern.colors, interactive = TRUE
)Exploration within each cluster
The example above plots all cells in one ternary plot, showing an overall view of the lineage relationship. However, closely placed clusters might overlap with each other, making it hard to distinguish the cells from different clusters. Furthermore, the velocity information is summarized within grids of the dot coordinates, which masks the difference of velocity between clusters closely placed. To address these issues, we can plot the cells from each cluster separately, and then the velocity arrows will only show the summary of included cells.
ternList <- plotTernary(
x = rna, clusterVar = annotation, vertices = vt.tern, features = gene.tern, veloGraph = velo,
byCluster = "all", dotColor = hspc.colors, labelColors = vt.tern.colors
)
# Remove legends that are redundant for the combined panels
ternList <- lapply(ternList, `+`, theme(legend.position = "none"))
# `patchwork` syntax for combining multiple plots
(ternList$`Prog DC` + ternList$Erythrocyte) / (ternList$HSC + ternList$MEP)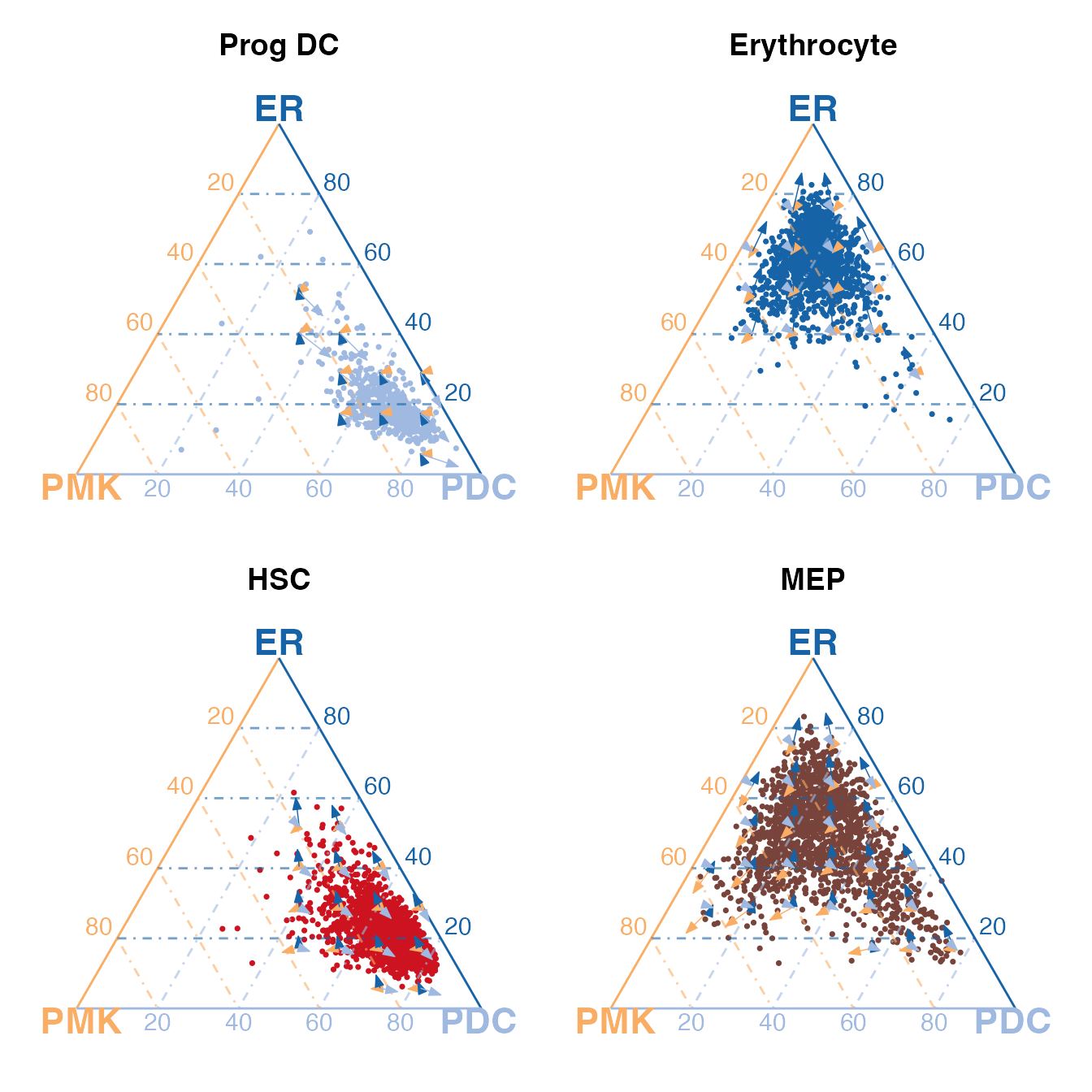
Above we show for single-cluster examples, including two terminal cell types ER and PDC, and two stem cell types HSCs and MEPs.
From the top row of subplots, we can see that the PDCs and ERs stay close to their corresponding vertices, respectively, and the differentiation potential is exclusively pointing to themselves, indicating that they are suitable for being set as the terminal cell types.
We can now clearly observe that HSCs stay close to the vertex of PDCs, while MEP cells diverge from the HSC and has a similar transcriptomic state to the ERs. Although we observe such similarities in transcriptomic states, we can also tell an obvious difference in the differentiation potential. The HSCs show differentiation potential to all vertices while PDCs are committed to themselves. Meanwhile, the MEPs are more inclined to differentiate into ERs and PMKs, which both belong to the myeloid lineage. These indicates that HSCs are more primitive and have the potential to differentiate into both lymphoid and myeloid lineages, while MEPs are more committed to the myeloid lineage.
Extending the 4th terminal fate with quaternary plots
Upon the visualization above, we further add another vertex for the granulocyte (GR) cluster to form a quaternary simplex.
vt.quat <- list(
PMK = "Prog MK",
ER = "Erythrocyte",
PDC = "Prog DC",
GR = "Granulocyte"
)
vt.quat.colors <- c(hspc.colors['Prog MK'],
hspc.colors['Erythrocyte'],
hspc.colors['Prog DC'],
hspc.colors['Granulocyte'])
gene.quat <- selectTopFeatures(x = rna, clusterVar = annotation, vertices = vt.quat, nTop = 30)plotQuaternary() is implemented for visualizing the
simplex in a 3D tetrahedron, allowing for exploring cell fates across
four terminal clusters. This function by default shows the simplex in a
plotly interactive view. A static image of specific perspective angle
can be shown with setting interactive = FALSE. Users can
scroll to zoom in and out, drag to rotate the 3D viewpoint, hover on the
dots or arrows for detailed information, and click on the legend tags to
toggle the visibility of the corresponding cluster or arrow.
plotQuaternary(
x = rna, clusterVar = annotation, vertices = vt.quat, features = gene.quat, veloGraph = velo,
dotColor = hspc.colors, labelColors = vt.quat.colors
)Granulocytes are known to be differentiated from granulocyte-monocyte progenitors (GMPs) instead of MEPs. The quaternary simplex plot clearly brings the GR cluster to a its new vertex, and shows that the GMPs are of different cell fates from MEPs.
PCA as an alternative feature input
This section demonstrates using PCA embedding as an input for creating ternary simplex plot. As compared to the top marker genes selected for the terminal clusters, PCA can potentially capture more global information of the dataset. Non-linear dimensionality reduction techniques such as UMAP or t-SNE are not recommended because they tend to distort the distances between cells and this does not match with the expectation of CytoSimplex’s modeling. Other methods such as Non-negative Matrix Factorization (NMF) shows great potential as well but will not be demonstrated in this tutorial.
We simply employ Seurat for preprocessing the raw count data and running PCA, leaving all parameters as default. Several points to note when using PCA as the input for CytoSimplex:
- The matrix must be transposed to proper direction such that each column is a cell.
- The argument
processedmust be set toTRUEto skip internal preprocessing on raw-count-alike data. - The distance metric calculation method is highly recommended to be
set as
"cosine"for PCA input.
library(Seurat)
hspc.pca <- CreateSeuratObject(counts = rna) |>
NormalizeData() |>
FindVariableFeatures() |>
ScaleData() |>
RunPCA() |>
Embeddings() |>
t()
sprintf("PCA: %s class, %d x %d", class(hspc.pca)[1], nrow(hspc.pca), ncol(hspc.pca))## [1] "PCA: matrix class, 50 x 11605"
plotTernary(
x = hspc.pca, clusterVar = annotation, vertices = vt.tern, veloGraph = velo,
processed = TRUE, method = "cosine",features = 1:30,
dotColor = hspc.colors, labelColors = vt.tern.colors, interactive = TRUE
)Similarly, we can generate a quaternary simplex plot with the PCA embedding.
plotQuaternary(
x = hspc.pca, clusterVar = annotation, vertices = vt.quat, veloGraph = velo,
processed = TRUE, method = "cosine", features = 1:30,
dotColor = hspc.colors, labelColors = vt.quat.colors, interactive = TRUE
)Compared to the plot above using top terminal marker genes, the PCA-simplex plot pushes the cells from the terminal clusters closer to their corresponding vertices, especially the PDCs. The MEP cluster is now placed with more similarity towards the PMK vertex while still being close to the ER vertex. This better matches to our expectation because the ER and PMK vertices are both in the myeloid lineage while MEP is on the progenitor trajectory of the myeloid lineage.